Project members

Project
Neurofibromatosis type 2 (MIM101000) is an autosomal-dominant disease with a frequency of 1/25000 livebirths. Patients are predisposed to develop bilateral vestibular schwannomas, although schwannomas in other cranial, spinal and peripheral nerves can also be observed. Additional clinical features include various type of nervous system tumors (meningiomas and ependymomas), ocular and cutaneous lesions. This tumor predisposition syndrome results from the loss of function of the NF2 tumour suppressor gene located on chromosome 22q12.2. Half of patients are sporadic cases and recent studies show the presence of mosaicism in 20 to 30% of de novo cases. Schwannomas are also observed in schwannomatosis (MIM162091) another tumor predisposition characterized by the development of multiple schwannomas. Germline mutations of SMARCB1 et LZTR1 genes localized in the proximity of the NF2 gene are involved in familial and sporadic forms of schwannomatosis.
We aim to understand the molecular bases of schwannomas development in NF2 and schwannomatosis by studying the functional consequences of genetic alterations on tumorigenesis using tumor samples, primary Schwann cells or immortalized Schwann cells genetically modified with CRIPSR Cas9 to selectively target NF2, LZTR1 and SMARCB1.
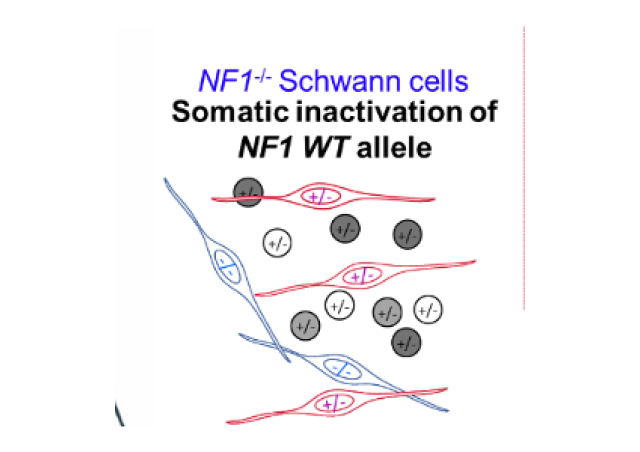
In accordance with results of other groups, we have recently shown that two thirds of NF2 patients’ schwannomas carry a germline mutation in the NF2 gene combined to the somatic inactivation of the second NF2 allele corresponding to the deletion of the NF2-SMARCB1-LZTR1 chromosomal region (Louvrier et al 2018).
Furthermore, schwannomas development in patients with a germinal mutation of SMARCB1 or LZTR1 are linked to the bi-allelic inactivation of these genes. This one can be associated with a mono or bi-allelic inactivation of NF2 (Kehrer et al 2017 for review). Finally, a recent analysis of the exomes of sporadic schwannomas has revealed the involvement of bi-allelic somatic mutations of NF2 in 77% of the tumors, often associated with mutations of LZTR1 (Agnihotri S et al 2016).
NF2 is primarily a major regulator of the Hippo signaling pathway. LZTR1 is a negative regulator of the Ras family of GTPases whereas SMARCB1 is involved in transcriptional regulation through chromatin remodeling. Although the main cellular functions of these three tumor suppressors are relatively well documented, the mechanisms of their functional synergies following co-inactivation are still poorly understood. On the other hand, it is likely that other cellular functions are involved in tumor development, particularly in the case of NF2. Interestingly, we observed that Merlin (encoded by NF2) modulates the dynamics of microtubule polymerization. This activity could explain our previous observations showing an accumulation of transmembrane growth factors receptors in NF2-mutated Schwann cells.
Our group has three main objectives: (1) to understand the functional consequences of the co-inactivation of NF2 and LZTR1 in Schwann cells, its impact on proliferation and transformation, its role in tumor development, and its consequences on the definition of potential therapeutic targets; (2) to determine the involvement of the regulation of microtubule dynamics in the tumor suppressor activity of NF2; (3) to develop a specific inhibitor of Yap and Taz, the end effectors of the Hippo signaling pathway. In collaboration with industry, we are evaluating the ability of short peptides to inhibit the transcriptional activity of Yap and Taz and specifically prevent the proliferation of NF2-mutated Schwann cells derived from animal models or isolated from human Schwannoma biopsies.



Funding