Project members





Project
NF1 is a tumor predisposition syndrome resulting from constitutional heterozygous mutations of the NF1 gene encoding neurofibromin, a RAS-MAPK pathway inhibitor. NF1 patients have an increased risk for malignant and non-malignant tumors compared with the general population. At present, no definitive treatment is available, and clinical management is typically limited to surveillance and symptomatic treatment, usually surgical, for specific complications. We aim to perform functional characterization of NF1 driven tumors, including genomic approaches in leukocyte and tumor samples from NF1 patients, and functional approaches in primary and tumor cell lines, and in genetically modified cells.

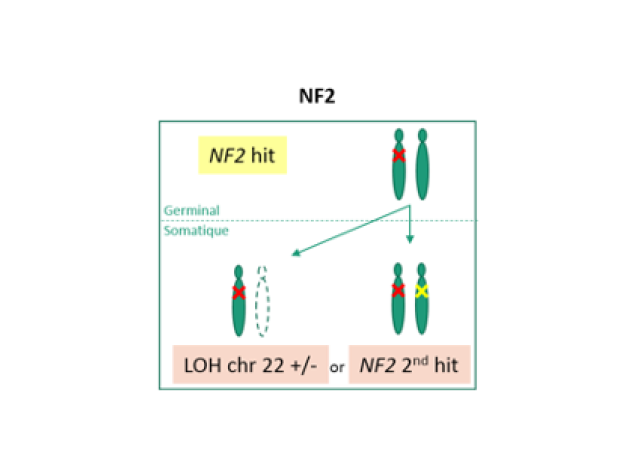
Neurofibromas are one of the major clinical features of NF1. They are benign peripheral nerve sheath tumors which consist in a proliferation of Schwann cells showing somatic inactivation of the NF1 WT allele. The malignant transformation of neurofibromas into MPNSTs (Malignant Peripheral Nerve Sheath Tumors) is the leading cause of death in NF1 patients. In 2014, we demonstrated for the first time that PRC2 (Polycomb Repressive Complex 2) played a key role in the development of NF1-associated MPNSTs, in an international collaborative study (De Readt et al. 2014). PRC2 is involved in maintaining transcriptional repression. EZH1/2, the catalytic PRC2 components, function as histone methyltransferase that di- and tri-methylates lysine 27 on histone 3 (H3K27me3). EED, SUZ12 and RBBP4/7 are required for complete function and stability of the PRC2 complex.
In collaboration with Raphaël Margueron’s team (Institut Curie), we aim to search for signaling pathways which are altered consequently to PRC2 mutations in MPNSTs. Spatial transcriptomics innovative approaches will be implemented to study the intra-tumoral heterogeneity of MPNSTs presenting mutations of the transcriptional repressor PRC2. In addition, we have selectively inactivated the PRC2 in Schwann cells immortalized from NF1-associated neurofibromas, using CRISPR Cas9 genomic editing techniques. We will use our genetically modified cell lines to ask how specific cell properties are affected by the lack of PRC2. By comparing PRC2 wild-type and mutant isogenic cell lines, these experiments should allow us to assess the contribution of PRC2 alteration to the malignant evolution of NF1 deficient cells, as well as the signaling pathways that could be targeted for therapy. In order to identify vulnerabilities associated with PRC2 mutations, two approaches will be used: chemical and genetic screens. We hope that these approaches will open new avenues to target MPNSTs. The same strategies will also be used to target the complete loss of function of the NF1 gene, that occurs in NF1-associated tumors.
In addition to nerve sheath tumors, our team is also interested in optic pathway gliomas associated with NF1. Our goal is to understand the molecular basis of glioma development in NF1 by studying the genetic alterations at the origin of tumor development.
Funding