Membres du projet





Projet
La NF1 est un syndrome de prédisposition tumorale causé par des mutations constitutionnelles hétérozygotes du gène suppresseur de tumeur NF1. Ce gène est localisé en 17q11.2 et code la neurofibromine, un inhibiteur de la voie RAS-MAPK. Les patients atteints de NF1 présentent un risque tumoral accru (tumeurs bénignes et cancers) et le traitement associé est généralement chirurgical et symptomatique selon les complications développées. Le développement de ces tumeurs à un impact sur la qualité de vie des patients mais également sur leur espérance de vie en cas de tumeurs malignes. Nous avons pour objectif d’étudier sur le plan fonctionnel les altérations génétiques à l’origine du développement tumoral à partir d’échantillons tumoraux, de cultures cellulaires primaires ou de lignées ou de modèles cellulaires génétiquement modifiés.

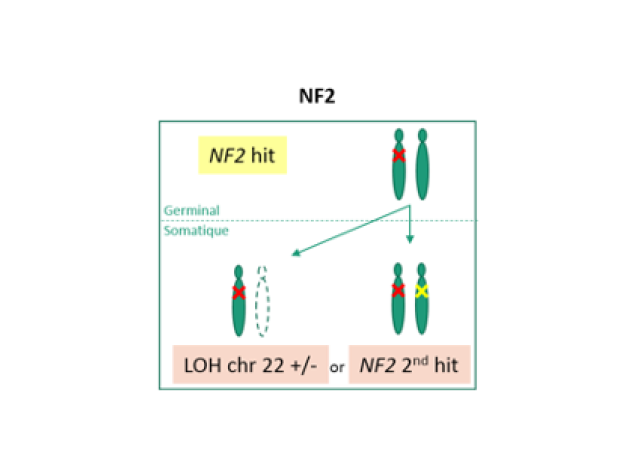
Les neurofibromes représentent une caractéristique clinique majeure de la NF1. Il s’agit de tumeurs bénignes des gaines des nerfs, composées notamment d’une prolifération de cellules de Schwann montrant une inactivation somatique de l’allèle sauvage du gène NF1. La transformation maligne de certains neurofibromes en MPNST (Malignant Peripheral Nerve Sheath Tumors) est la principale cause de surmortalité chez les patients atteints de NF1. En 2014 dans le cadre d’une collaboration internationale (De Readt et al., 2014), nous avons montré pour la première fois que le PRC2 (Polycomb Repressive Complex 2) jouait un rôle majeur dans le développement des MPNST associés à la NF1. Le PRC2 est impliqué dans le maintien de la répression transcriptionnelle ; EZH1/2, les sous-unités catalytiques du complexe, sont des méthyltransférases responsables de la di- et tri-méthylation de la lysine 27 de l’histone 3 (H3K27me2/3). Les protéines EED, SUZ12 et RBBP4/7 sont nécessaires à la stabilité et à la fonction du PRC2.
Nous avons pour objectifs d’identifier les voies de signalisation altérées suite aux mutations perte de fonction du PRC2 dans les MPNST, en collaboration avec l’équipe de Raphaël Margueron (Institut Curie). Des approches innovantes de transcriptomique spatiale seront mises en œuvre pour l’étude de l’hétérogénéité intratumorale des tumeurs malignes des gaines nerveuses présentant des mutations du répresseur transcriptionnel PRC2. De plus, par des techniques d’édition génomique CRISPR Cas9, nous avons sélectivement inactivé le PRC2 au sein de cellules de Schwann immortalisées à partir de neurofibromes associés à la NF1. Nous employons ces modèles génétiquement modifiés pour identifier les propriétés cellulaires modifiées par l’inactivation du PRC2. La comparaison des cellules isogéniques sauvages et PRC2 mutées, nous permettra d’évaluer la contribution des altérations du PRC2 à la transformation maligne des cellules de Schwann NF1 déficientes, ainsi que les voies de signalisation qui pourraient être ciblées par des approches thérapeutiques. Pour identifier des vulnérabilités associées aux mutations perte-de-fonction de NF1 et du PRC2 accompagnant le développement tumoral, des approches de cribles pharmacologiques et génétiques seront mises en œuvre sur les différents modèles isogéniques que nous avons développés.
Outre les tumeurs des gaines nerveuses, notre équipe s’intéresse également aux gliomes des voies optiques associés à la NF1. Nous avons pour objectif de comprendre les bases moléculaires du développement des gliomes dans la NF1 en étudiant les altérations génétiques à l’origine du développement tumoral, à partir d’échantillons tumoraux.
Financements