Project members
Project
Our aim is to study the mechanisms of neutrophil activation and the role of proteinase 3 in autoimmune necrotizing vasculitis and especially in granulomatosis with polyangitis (GPA, formerly called Wegener’s granulomatosis).
The pathogenesis of anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitides is poorly understood but it is consistent with a pivotal role for neutrophils since they are both effector cells responsible for endothelial damage and targets of autoimmunity.
Systemic vasculitides include a heterogeneous group of systemic diseases characterized by vascular necrosis and perivascular inflammation in small vessels. Notably, the clinical presentation of these diseases is different and varies according to the localization and the vessel size involved. ANCA-associated vasculitides involve small vessels and comprise microscopic polyangiitis (MPA), Churg-Strauss syndrome and granulomatosis with polyangitis (GPA, formerly called Wegener’s granulomatosis). In both microscopic polyangiitis and Churg-Strauss syndrome, myeloperoxidase is the main target antigen, whereas Wegener’s granulomatosis is strongly associated with ANCA directed against proteinase 3 (PR3).
ANCA-associated vasculitis are characterized by systemic inflammation associated with an immune dysregulation and an autoimmunity targeting neutrophils. In granulomatosis with polyangitis (GPA), neutrophils strongly express membrane-associated proteinase 3, which constitutes a pro-inflammatory factor. Although proteinase 3 appears to play an instrumental role in Wegener’s granulomatosis pathophysiology, its functions in neutrophils are not well understood.
Physiopathology of ANCA-associated vasculitis: role of protéinase 3
It is classically accepted that, during vasculitis, an infection will trigger a "pre-activation of neutrophils" resulting in the secretion of inflammatory cytokines such as TNF-alpha and an increased expression of adhesion molecules. Under these conditions, neutrophils express granular proteins on their surface, in particular ANCA targets such as myeloperoxidase or proteinase 3, which can then bind anti-MPO or anti-PR3 ANCAs, and activate neutrophils. These neutrophils will then release their endothelium-damaging mediators such as oxidants and proteases, which cause vascular damage. The accumulation of activated and apoptotic neutrophils suggests a failure to resolve the inflammation.
We have shown that apoptotic neutrophils from patients with GPA express membrane PR3 and that it underlies a defect in phagocytosis by macrophages and disruptions in innate and adaptive immunity (Mouthon et al 2012 Medical Press; Millet et al. Ann Rheum Dis. 2013).
Our Project is focused on the structural and functional analysis of the autoantigen PR3 to specifically target particular domains to alter its functions.
Proteinase 3 has the typical fold of chymotrypsin-like serine proteases (Hajjar et al. FEBS J. 2010): two beta-barrels made each of six anti-parallel beta-sheets (purple) and a C-terminal alpha helix (yellow) (Figure 1). The active catalytic triad site is located between the two barrels (his71, asp118 and ser203 in green). The substrate (red) is positioned optimally in the active site. PR3 has an hydrophobic patch to insert into the membrane (Hajjar et al Proteins 2008) (shown in yellow and orange) (Molecular modeling was performed by the group of Nathalie Reuter, University of Bergen ; Hajjar et al Proteins 2008).
PR3 could bind to phosphatidylserine expressed on apoptotic cells or on microvesicles to further modulate and disseminate inflammation (Martin et al J Biol Chem, 2016).

The structural studies on PR3 from the team have provided evidence that PR3 was associated with membrane proteins at the surface of apoptotic neutrophils. PR3 is a phosphatidylserine–binding protein can modulate the function of proteins involved in the recognition of apoptotic cells including the phospholipidscramblase 1, calreticulin, the C1q fragment of the complement and annexine-A1. The presence of PR3 in the proteic platform seems to participate to the pathogenesis of renal vasculitis (Figure 2).

Disturbed death pathways in neutrophils from GPA patients
The functional study of neutrophils from GPA patients in the NEUTRO-VASC clinical study (PH-RC national) was carried out to examine whether measurement of neutrophil activation could be used as biomarkers in this disease.
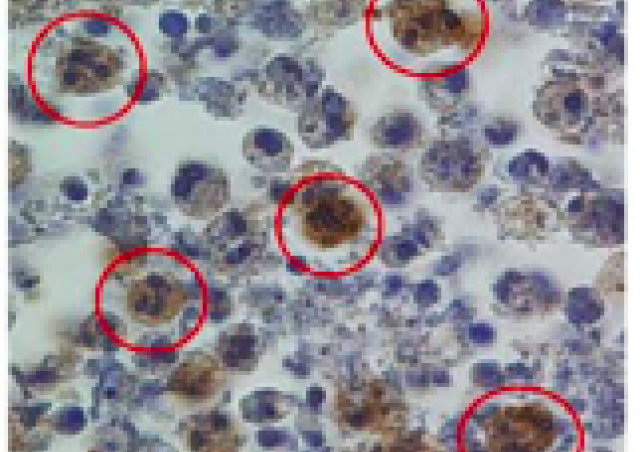
Proteomic analysis of neutrophil cytosol in more than 70 patients with GPA has shown that neutrophils from patients have an intrinsic dysregulation of death pathways and in mechanisms of apoptotic cell recognition thereby explaining the defect in the resolution of inflammation observed in this diseases (Figure 3).
More importantly, a network of membrane proteins containing the PR3 autoantigen, annexine-A1 and phospholipidscramblase1 was expressed at the surface of apoptotic neutrophils only in GPA patients with a renal involvement suggesting that these proteins were linked to the clinical state and renal disease.


Mechanisms of immune dysregulation in ANCA-associated vasculitis: role of the autoantigen proteinase 3.
ANCA-associated vasculitis are characterized by systemic inflammation associated with an immune dysregulation and an autoimmunity targeting neutrophils. To date, the cause of these diseases are unknown.
Our research project is aimed at understanding the role of neutrophils in the immune dysregulation in GPA and more specifically the role of the traget autoantigen PR3.
In 2015, the team has discovered a novel pathway by which PR3 expressed on apoptotic neutrophils could initiate a cascade of proinflammatory reaction and favor autoimmunity. This study show for the first time that autoimmune vasculitis could have an autoinflammatory component involving interleukin-1 beta.
Proteinase 3 (PR3) is stored into the azurophil granules of circulating neutrophils. During vascular inflammation, neutrophils are activated by ANCA and undergo apoptosis. During this process, they can express PR3 at the plasma membrane, which can interfere with phagocytosis (Kantari et al Blood 2007) and activate macrophages through the MYD88/interleukin 1 pathway inducing the production of inflammatory cytokines and chemokines (Gabillet et al J Immunol 2012). PR3 mimics a danger signal for macrophages resulting in a microenvironment favouring activation of pDCs, which are key cells in the immune silencing associated with the phagocytosis of apoptotic cells. Phagocytosis of apoptotic cells expressing PR3 results in an inhibition of the generation of regulatory T cells and a polarization of CD4 positive T helper cells into a Th9 profile. In addition, anti-PR3 ANCA further enhances the generation of Th17 cells thus potentiating inflammation. Generation of G-CSF potentiates PR3 synthesis in myeloid precursors leading to increased PR3 expression in mature neutrophils, and thus in turn potentiating inflammation (Figure 4).
Funding