Members
Project
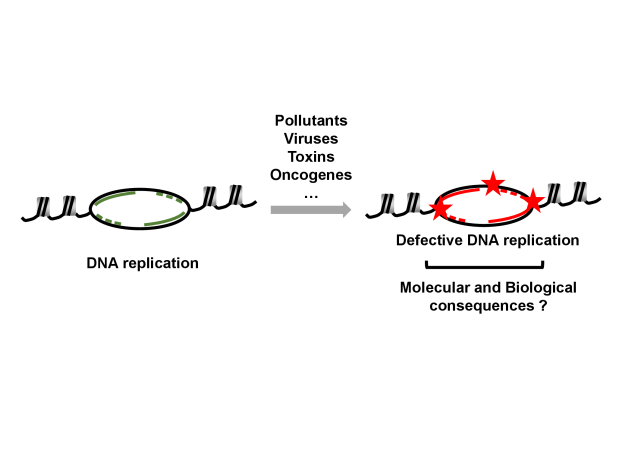
Several human syndromes are caused by mutations in genes coding for factors that regulate DNA replication: Seckel syndrome, Bloom syndrome, to name but a few. The common feature of these syndromes is a growth defect or primary dwarfism.
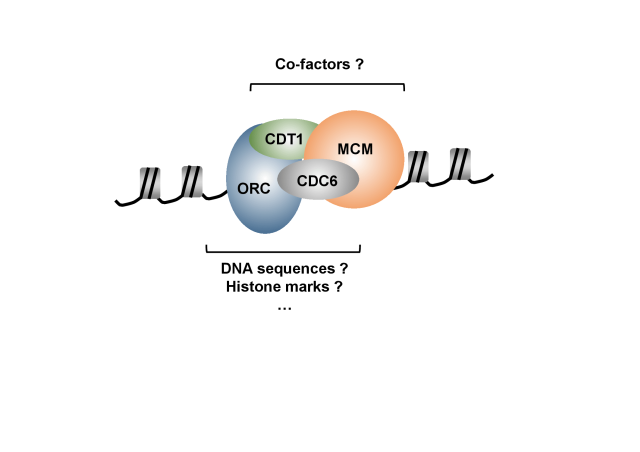
Meier-Gorlin Syndrome (MGS) is a rare autosomal recessive disorder that combines short stature with poor growth with a variable number of developmental abnormalities such as small ears or absent patella (ORPHA: 2554). Since 2011, research has shown that mutations in ORC (origin of replication recognition complex) subunits, CDT1, GEMININ, CDC45 and MCM complex subunits are involved in the etiology of the disease. The mutations causing the most severe phenotype alter the function of the gene encoding the ORC1 subunit of the ORC complex, and the most recurrent of them corresponds to the substitution of arginine 105 by glutamine in the BAH (for "bromo-adjacent homology") domain, a highly conserved functional domain in mammals (OMIM: 224690).
Our team seeks to understand :
- What are the molecular and cellular consequences of MGS mutations in human cells?
- Are there other, non-canonical functions associated with replication initiation factors?
- What are the biological functions of replication initiation factors during early embryogenesis?
To answer these questions, the team has several cell models derived from patient samples, such as fibroblasts and lymphocytes, as well as a mouse model of the disease. We are also actively collaborating with the physicians of the 'Filière Santé Maladies Rares OSCAR'.
Fundings