

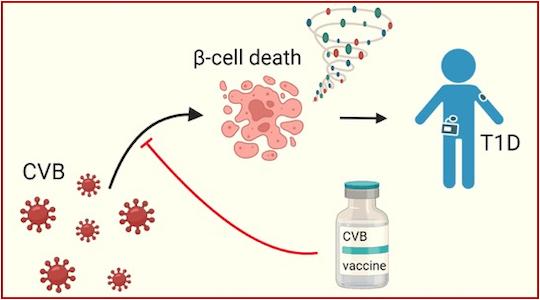
Despite the lack of evidence for a causal relationship, CVB infections are recognized as plausible environmental triggers for T1D, due to their temporal correlation with the appearance of autoantibodies against beta cells (the first measurable signs revealing autoimmunity), and the detection of the virus in the pancreas of diabetic patients. Additionally, CVB can directly and efficiently infect beta cells, leading to their death. These observations led to the development of a PRV-101 vaccine against CVB, which was the subject of a pilot clinical trial with promising results in terms of safety and induction of protective antiviral antibodies. Before developing trials on a larger scale, it appeared important to better understand the immunity induced by CVB. While antibody responses have been widely studied, those of CD8+ T cells (lyT8), central players in antiviral responses, as well as autoimmune responses against beta cells, were largely unknown.
The researchers first infected beta cells in vitro with CVB to study surface-exposed viral antigens for immune recognition of infected cells. These experiments showed that CVB “hides” very effectively inside beta cells, exposing few antigens. Additionally, CVB passes from one beta cell to another using cell protrusions, further avoiding immune recognition.
In a second step, the viral antigens were used as molecular “baits” to detect circulating lyT8s that could recognize them in individuals who had been infected in the past. These experiments revealed a second surprise: the lyT8 response to CVB is very weak, and does not lead to the formation of an effective immune “memory” (capable of protecting the individual in the event of reinfection).
The third step consisted of isolating these lyT8s to bring them into contact in vitro with infected beta cells. These experiments showed that beta cells were mainly destroyed by CVB, but spared by antiviral lyT8 (though supposed to kill infected cells in order to limit the spread of the infection).

These results are important because, before this study, two scenarios could explain how CVB triggers T1D. First, weak anti-CVB immune responses would promote viral spread and persistence, thereby leading to beta cell destruction caused primarily by the virus itself. Second, strong antiviral immune responses could lead to destruction of infected beta cells by antiviral lyT8s recruited following infection. If in both cases the end result is the release of autoantigens from the beta cell in a context of inflammation and the triggering of the T1D autoimmune response, the implications for vaccination strategies are different. Indeed, anti-CVB vaccination would be useful to strengthen weak immune responses and thus limit viral spread, but potentially dangerous if it were to lead to an increased antiviral response against beta cells.
These results demonstrate a primary and direct destructive effect of CVB on beta cells, favored by weak antiviral immune responses. It is therefore relevant to try to strengthen these responses through vaccination, which could provide a primary prevention strategy for T1D, by intervening on the triggering factors even before the onset of autoimmunity. Furthermore, these results explain how a widespread infection like CVB leads to T1D in only a few individuals: it is not the infection itself that is to blame, but the more or less effective reaction of the immune system against this infection which, if faulty, favors the spread of CVB to beta cells and their destruction.
These studies are now continuing as part of a European ENT1DEP network (www.ent1dep.eu) to provide new means of early detection of a risk of developing T1D following CVB infection and to continue the clinical development of the vaccine. A viral origin is also suspected for other autoimmune diseases, for example multiple sclerosis, this research could broaden its relevance to the field of autoimmunity in the broad sense.
Reference
Federica Vecchio et al. Coxsackievirus infection induces direct pancreatic β cell killing but poor antiviral CD8+ T cell responses. Sci. Adv.10,eadl1122(2024). DOI:10.1126/sciadv.adl1122



