L’objectif principal de notre équipe est d’obtenir une meilleure compréhension des mécanismes moléculaires, cellulaires et biochimiques qui régulent le processus d'initiation de la réplication de l'ADN et le maintien de l'intégrité des chromosomes lors de la division cellulaire.
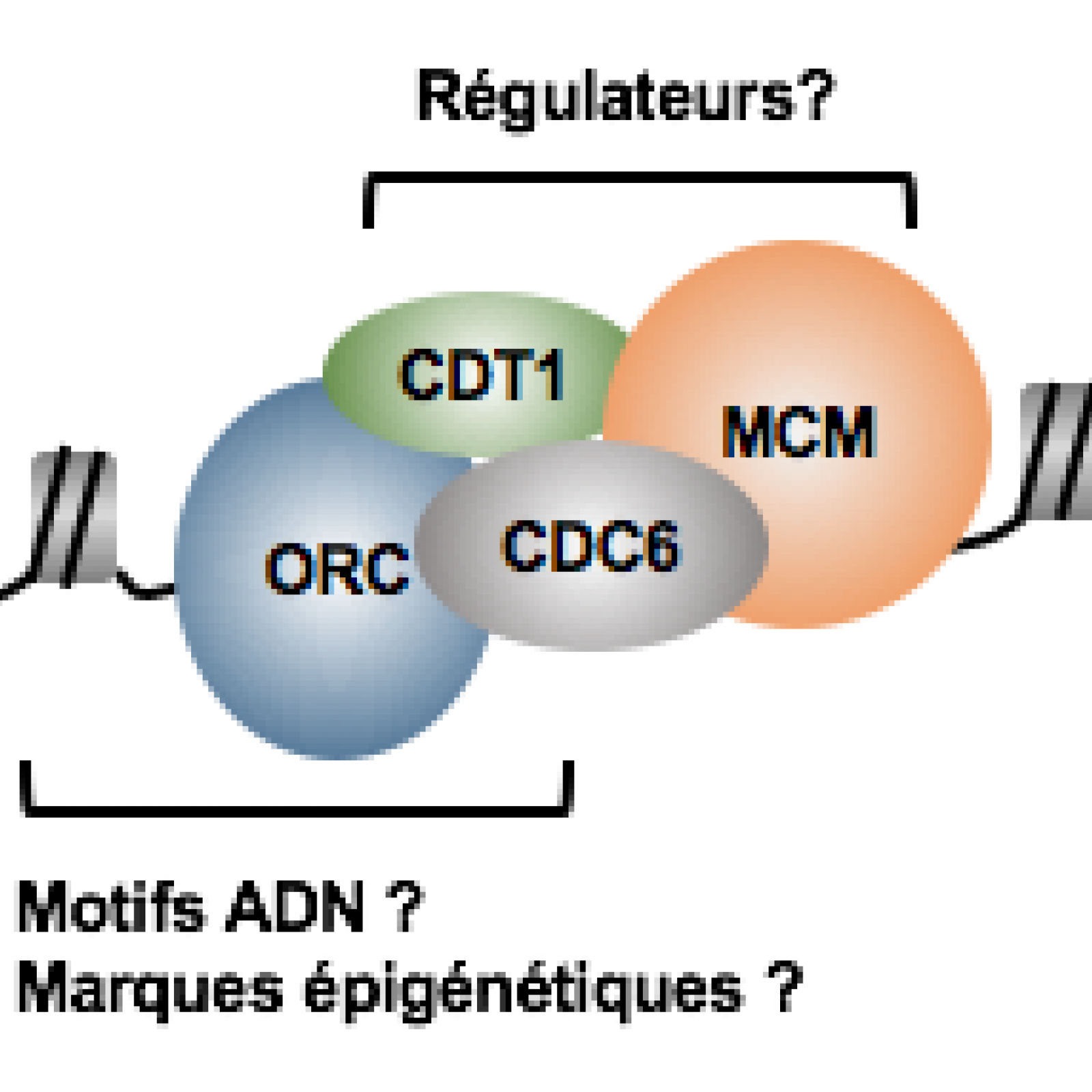
L'initiation de la réplication du génome débute par l'assemblage d'un complexe macro-moléculaire appelé le complexe de pré-réplication au niveau de milliers d’origines de réplication au cours de la phase G1 du cycle cellulaire. C'est à partir de ces origines de réplication que s'initie la réplication de l'ADN et que progresse dans les deux sens les fourches de réplication.
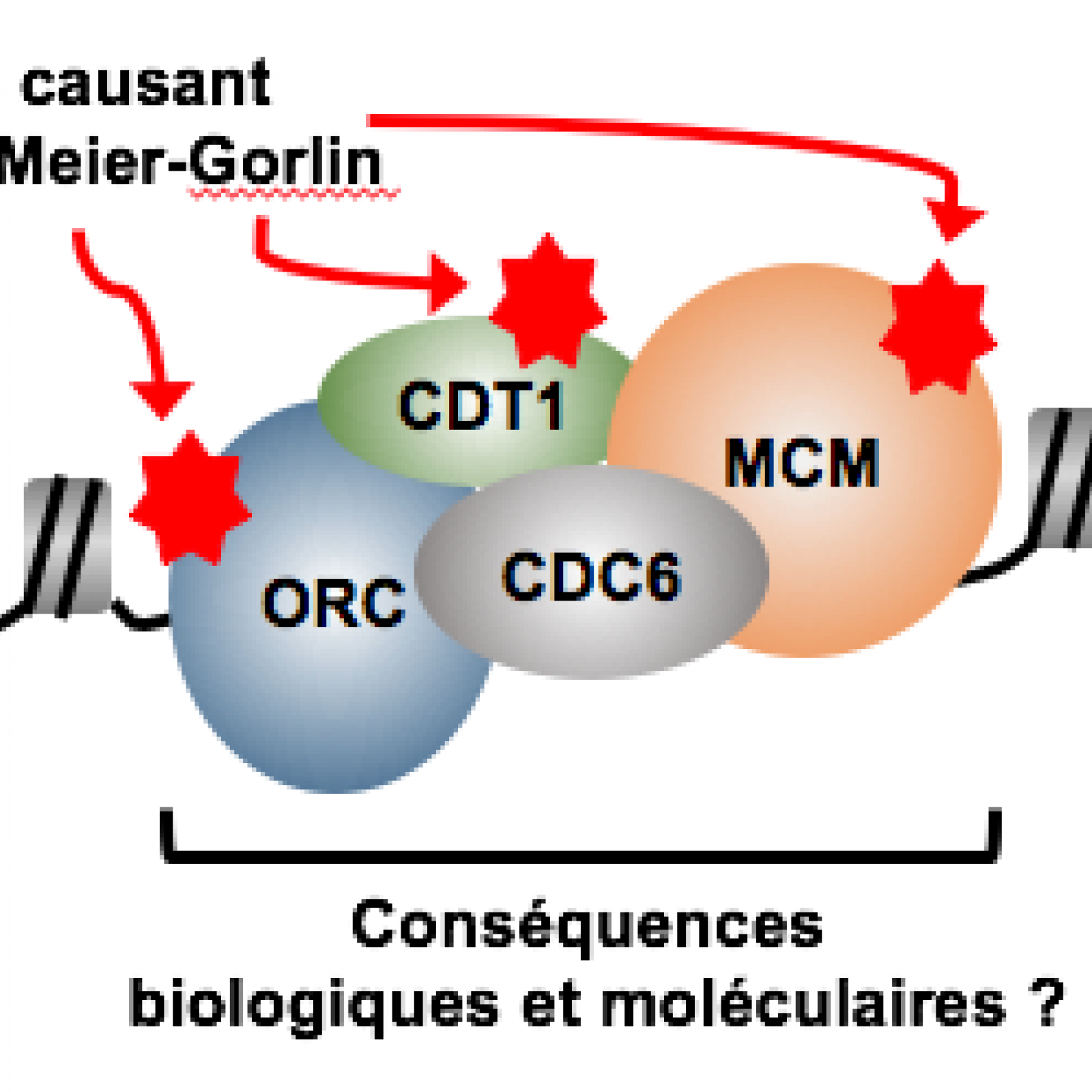
Notre équipe développe des approches originales de génomique, biologie moléculaire et biologie cellulaire pour obtenir une meilleure compréhension des acteurs et des régulateurs essentiels à la formation et la fonction de ce complexe de pré-réplication et des fourches de réplication. A l'heure actuelle nous nous intéressons principalement aux rôles de plusieurs facteurs de transcription et de remodelage du paysage chromatinien dans ce contexte. Nous étudions par ailleurs les conséquences qu'une perturbation du processus d'initiation de la réplication peut avoir sur le devenir d'une cellule, et l'implication possible dans l'émergence de certaines pathologies, comme le cancer et le syndrome de Meier-Gorlin.
L'activité scientifique de l'équipe s’intègre aux axes thématiques « cancer » et « plasticité génétique et cellulaire » de l'institut, ainsi qu'aux axes de recherche du laboratoire d’excellence « Who am I ? » et de la filière santé maladies rares de l'os, du calcium et du cartilage « OSCAR », auxquels les membres de l'équipe sont associés.